Is AZT a DNA chain terminator?
A good scientist values criticism even higher than friendship; no, in science criticism is the height of friendship
Francis Crick
In the package insert provided with its AIDS drug AZT, GlaxoSmithKline makes the claim: ‘Zidovudine [AZT] is an antiviral agent active in vitro against retroviruses including the Human Immunodeficiency Virus (HIV).’ And then goes on to explain how: ‘Zidovudine is phosphorylated in…cells to the…triphosphate (TP) derivative… The formation of further proviral DNA is blocked by incorporation of zidovudine-TP into the chain and subsequent chain termination.’
In other words, GSK claims that AZT is a DNA chain terminator. Dr Peter Duesberg, Professor of Molecular Biology at the University of California at Berkeley, and arch-critic of the drug, agrees. So does pharmaceutical biochemist Dr David Rasnick, a visiting scientist in his laboratory. Their paper The AIDS dilemma: drug diseases blamed on a passenger virus, published in Genetica in 1998, contains a valuable critique of AZT’s toxicity. It describes the drug as a ‘chain terminator…synthesized over thirty years ago for chemotherapy. Since the operating principle of cancer chemotherapy is to kill growing cells by terminating cellular DNA synthesis at micromolar concentrations, AZT was predictably pathogenic’. What Duesberg and Rasnick mean by ‘chain terminator’ is explained in an earlier paper that Duesberg co-wrote with David Chiu, also (then) of Berkeley, The toxicity of azidothymidine (AZT) on human and animal cells in concentrations used for antiviral therapy, published in the same journal in 1995: ‘AZT…is an analog of thymidine in which the 3’ hydroxyl group is replaced by an azido group. This prevents the extension of a growing DNA strand ending with AZT to the five prime end of another nucleotide triphosphate. Thus AZT functions as a terminator of DNA synthesis.’ Rather clumsily put, because they imply the punchline instead of stating it expressly. Slot the following sentence in, after their first, and you’ll get it: ‘The drug is incorporated into growing DNA in place of thymidine triphosphate.’ Duesberg makes this implication clear in his popular book Inventing the AIDS Virus, describing the putative chain terminating action of AZT like this: ‘Every time a cell divides, it must copy its complete genetic code, allowing one copy for each new cell. Genetic information is stored as a sequence of four “letters” in long chains of DNA, known as chromosomes. [Of course there is a lot more to chromosomes than DNA.] Each building block of DNA is linked to the one before it, almost like train cars. … [AZT] surreptitiously enters the growing DNA chain while a cell is preparing to divide and acts as a premature chain “caboose”, blocking further DNA building blocks from being added.’ But the fallacy at the root of that analogy is the notion that DNA grows like an ever-extending row of railway cars or a string of pearls. Because that’s a textbook fairy story, and not how it really happens at all. And whether AZT actually slips into human (or viral) DNA as claimed, is the very subject of this article.
This pearl necklace notion is a popular one; drug specialists in the US FDA propound it too. A correspondent of mine, Gerrit Brand, a theologian with a PhD from the University of Utrecht, asked them: ‘How can AZT be an antiretroviral drug if it is not triphosphorylated in human cells to the inhibition concentration required to act as a DNA chain terminator?’ On 28 February 2002 he got his reply. ‘Thank you for your message to the Center for Drug Evaluation and Research (CDER), one of the five centers within the Food and Drug Administration (FDA). … Below is the response we’ve received from the Division of Antiviral Drug Products: “We do not understand the statement that AZT is ‘not triphsophorylated in human cells to the inhibition concentration required to act as a DNA chain terminator.’ There is really no concentration ‘required’ for triphosphorylated AZT (AZTTP) to act as a chain terminator. AZTPP competes with triphosphorylated thymidine (TTP) and inhibits reverse transcriptase (the enzyme that copies RNA into DNA). Whenever even one molecule of AZT is incorporated into the DNA chain, the chain is terminated.”‘ An indication of that fallacy (and Rasnick, we’re about to see, would later repeat it) lies in the fact that according to Duesberg and him in The AIDS Dilemma, 500 mg of AZT (a typical medical dose) comprises 1021 molecules: 107 molecules of AZT for every cell of our bodies. If even just one in a thousand AZT molecules was triphosphorylated inside our cells, and then slipped into growing human DNA chains and terminated them, nobody on AZT would last much longer than a man dangling on a rope.
GSK claims further: ‘Zidovudine-TP acts as an inhibitor of, and substrate for, the viral reverse transcriptase’ and ‘Competition by zidovudine-TP for HIV reverse transcriptase is approximately 100-fold greater than for cellular DNA polymerase alpha.’
What GSK means by this claim is that AZT terminates HIV DNA specifically, and not human DNA – or at least hardly any. Duesberg, Rasnick, and Chiu part company with GSK on this score. They contend that AZT terminates cellular DNA too. Decimating cells as it does so. And that this is why AZT is so very poisonous.
The notion that DNA chain termination is the principle activity of AZT has its origin in a speculation mooted by Furman and others, including researchers in the manufacturer’s employ, in their paper3’-Azido-3’-Deoxythymidine Triphosphate as an Inhibitor and Substrate of Purified Human Immunodeficiency Virus Reverse Transcriptase published in Antimicrobial Agents and Chemotherapy in 1987. They proposed: ‘Incorporation of azidothymidylate [AZTTP] into a growing [proviral HIV] DNA strand should terminate DNA elongation and thus inhibit DNA synthesis.’ On the basis of this tentative model GSK markets AZT as an anti-HIV drug. Although reading their package insert you’d think that AZT terminates DNA as an established fact.
On 1 September 1999 the DNA chain termination model for both AZT’s alleged pharmacological action (per GSK) and toxic ill-effects (per Duesberg, Chiu and Rasnick) was meticulously examined and disproved in a monumental literature review: A Critical Analysis of the Pharmacology of AZT and its Use in AIDS by Australian medical physicist Eleni Papadopulos-Eleopulos and others, published in Current Medical Research and Opinion, 15, Special Supplement. The paper concluded: ‘Based on all these data it is difficult if not impossible to explain why AZT was introduced and still remains the most widely recommended and used anti-HIV drug. [The continued administration of AZT] either alone or in combination…to HIV sero-positive or AIDS patients warrants urgent revision.’ The paper caught the eye of the world’s most prestigious science journal, Nature, which noted the paper with a growl, but didn’t address its enormous implications. GSK simply ignored it and continued marketing AZT as an anti-HIV drug.
Early in 2001 I began preparing particulars of claim for damages for loss of support and psychic trauma suffered by the widow and child of an attorney, James Hayman, who had succumbed to its toxicities. The lynchpin of the claim was the Papadopulos-Eleopulos critique. I thought it useful to elicit GSK’s response to the points it made, not because I had any doubts about them, but because I was curious to draw the company out before instituting the action. So I wrote an Open Letter to John Kearney, Chief Executive Officer of GSK in South Africa, incorporating a table presenting the triphosphorylation data that I received from Papadopulos-Eleopulos and Turner (which I extended to incorporate subsequent studies), and their ‘viral load’ graph that they had provided me. I sent it to the South African investigative journal noseweek, which in turn passed it on to GSK for a response. This is the letter:
In the package insert supplied with AZT you allege that it’s converted by enzymes inside human cells from its parent form as a pro-drug into its active agent, AZT triphosphate. And that AZT triphosphate stops HIV replication by being incorporated into growing proviral DNA chains during the reverse transcription of HIV RNA.
In November 1986 Furman and others including researchers from Wellcome Research Laboratories (a division of your company in an earlier incarnation) reported their finding that in the most ideal artificial conditions in vitro, the minimum concentration of AZT triphosphate necessary to inhibit proviral HIV DNA chain synthesis significantly, i.e. have an antiretroviral effect, is 0.7mM (1).
But sixteen studies conducted since then have found that in the real world in vivo, our cells can’t triphosphorylate AZT to anything like that level, with the best of the studies reporting AZT to be triphosphorylated in cells in vivo at levels one, even two orders of magnitude below the drug’s minimum effective concentration (2).
Would you please explain then why you persist in claiming that ‘Zidovudine [AZT] is phosphorylated in...cells to...the triphosphate (TP) derivative...’ – by implication to effective virustatic concentrations in vivo – when study after study has consistently shown that it isn’t?
If AZT prevents HIV replication by terminating proviral HIV DNA chain synthesis as you claim, one would expect the medicine to result in a consistent, sustained and simultaneous fall over time in all direct markers conventionally considered to indicate HIV infection levels, namely HIV DNA (viral burden), HIV RNA (viral load), detection of p24 and reverse transcriptase (viral isolation), and p24 antigenaemia. But all reported studies of the effect of AZT on these parameters in vivo show that the drug has no such anti-HIV effect (3). None at all on HIV DNA synthesis (viral burden), which flatly refutes your key claim that the drug blocks it. An insignificant effect on HIV RNA (viral load) (4). And none on the rest. All of which is perfectly predictable, since AZT is triphosphorylated by our cells negligibly, as we’ve seen.
So why do you still claim that ‘Zidovudine-TP acts as an inhibitor of, and substrate for, the viral reverse transcriptase’, that ‘The formation of further proviral DNA is blocked by incorporation of zidovudine-TP into the chain and subsequent chain termination (sic)’, and that AZT is thus ‘an antiviral agent ... active against ... HIV’, when all studies of the effect of AZT on direct markers for HIV infection levels in vivo have demonstrated these claims to be untrue?
And why do you continue to claim that AZT is ‘effective’, when the only long term, large scale, prospective, randomised, double-blind, placebo-controlled, clinical AZT study yet conducted – the Concorde trials in England, Ireland and France reported in 1994, involving 1749 symptom-free HIV-positive individuals – found that AZT has no therapeutic benefits when administered early (5), and the extended results of the study a year later showed ‘a significant increased risk of death among the patients treated early’ (6) ?
In your reply addressing the triphosphorylation and efficacy issues that I’ve raised, please leave out the effect of AZT on T4 (CD4+) cell counts – a non-specific, indirect marker modulated by cell poisons like AZT independently of any antiviral activity (7). The same goes for antibody levels.
(1) Proceedings of the National Academy of Sciences of the United States of America 1986; 83: 8333-7
(2) See table below. [Annexure ‘B’ of Hayman particulars of claim]
(3)Current Medical Research and Opinion, 1999; Volume 15; Special Supplement – posted online at: www.librapharm.co.uk/cmro/vol_15/supplement/main.htm and www.virusmyth.net/aids/data/epazt2.htm
(4) See graph below
(5) Lancet 1994; 343:871-81 (6) New England Journal of Medicine 1997; 336:958-9 (7) AIDS 1996; 10:1444-1445
Changes of HIV viral load induced by AZT
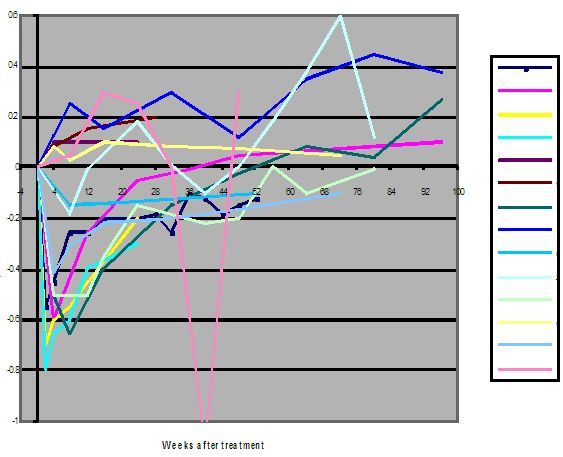
a) Eron JJ et al. NEJM 1995;333:1662-9
b) De Jong MD, et al. PNAS 1996;93:5501-6
c) Katlama C, et al. JAMA 1996;276:118-25
d) Katlama C, et al. JAMA 1996;276:118-25
e) Staszewski S et al. JAMA 1996;276:111-7
f) Carr A, AIDS 1996;10:635-41
g) O’Brien WA, et al. NEJM 1996;334;426-31
h) O’Brien WA, et al. NEJM 1996;334;426-31
i) Katzenstein D, et al. NEJM 1996:1091-8
j) Bakshi SS, et al. J Infect Dis 1997;175:1039-50
k) Bruisten SM et al. AIDS Res & Hum Retr 1998;12:1053-8
l) Delta Committee. AIDS, 1999:57-65
m) Delta Committee. AIDS, 1999:57-65
n) Lillo FB, et al. AIDS 1999;13:791-6
1. According to American HIV experts Saag, Shaw and Coombs and their associates in their article HIV viral load markers in clinical practice in Nature Medicine 1996; 2(6): 625-9: ‘A three-fold or greater sustained reduction (>0.5 log) of the plasma HIV RNA levels is the minimal response indicative of an antiviral effect... [R]eturn of HIV RNA levels to pre-treatment values (or to within 0.3 – 0.5 log of the pre-treatment value), confirmed by at least two measurements, is indicative of drug failure’.
2. According to the 1997 British HIV Association guidelines for antiretroviral treatment published in Lancet 1997; 349:1086-1092: ‘If the viral load has not fallen by about 1 log 8-12 weeks after treatment initiation consideration should be given to modify therapy’.
3. All studies in which the effect of AZT on HIV viral load in patients has been investigated, have consistently established that AZT taken alone or in combination with other drugs is not able to induce a sustained decrease in the ‘plasma HIV RNA level’ of >0.5 log (the American criterion for anti-HIV drug efficacy), much less 1 log (the British criterion).
4. By both the American and British criteria mentioned above, AZT fails to achieve ‘the minimal response indicative of an antiviral effect’ and is therefore a ‘drug failure’ i.e. ineffective as an antiviral medicine against HIV.
I reckoned my Open Letter would interest the AIDS dissident crowd, so I sent a copy to Rasnick in April 2001 for distribution via his mailing list. He circulated my Open Letter as requested, but under this disclaimer:
I have a small comment regarding the Perth Group’s arguments that AZT is not sufficiently triphosphorylated in vivo. I generally avoid technical discussions when it comes to AIDS but I feel that it is necessary here. AZT was not designed nor intended by its inventor to be an inhibitor of any enzyme. AZT and the other DNA chain terminating drugs were designed to be substrates that mimic natural nucleosides that get triphosphorylated and are then added to growing chains of DNA. The purpose of DNA chain termination is to kill the cell. That’s just what happens. For example, below is a recent paper that actually measured AZT incorporated into the DNA of humans taking the drug.
From O. A. Olivero et al. in their paper entitled ‘Incorporation of zidovudine into leukocyte DNA from HIV-1-positive adults and pregnant women, and cord blood from infants exposed in utero’ (1999) AIDS 13: 919-925:
“We show here that [AZT] is incorporated into leukocyte DNA of most individuals receiving [AZT] therapy, including infants exposed to the drug in utero. Further study of the biological consequences of [AZT]-induced DNA damage in the human population is warranted.”
Whatever efficiency there is of AZT triphosphorylation in vivo, it is certainly sufficient to allow AZT to be incorporated into growing DNA chains. DNA chain termination is a serious thing for a cell to experience. It doesn’t require very much incorporation of a DNA chain terminator to kill a cell. To inhibit an enzyme is a different matter. It generally takes concentrations 10-fold greater than the inhibitor’s Ki value to be effective in vivo. That means that you need at least10 nM concentration of inhibitor to be effective if the Ki of the inhibitor is 1 nM. Biology being what it is, typically you need even higher concentrations of inhibitor to be effective. But low efficiency drugs can be very toxic, nevertheless. For example, mitomycin C is a very toxic substance even though it is not a very efficient drug. Mitomycin C generally makes only one or two DNA chain cross-links per cell. But that is all that is needed to kill the cell. I accept that AZT is an insane drug to give anybody. First, because it cannot, even in principle, benefit AIDS patients because HIV does not cause AIDS. Second, AZT is well documented to be very toxic and is lethal if taken long enough. See for example our paper “The AIDS dilemma: drug diseases blamed on a passenger virus”, Duesberg, P. H. and Rasnick, D. (1998) Genetica 104, 85-132.
Not surprisingly, fellow researcher at the US National Cancer Institute and coauthor of the study led by Dr Ofelia Olivero that Rasnick relied on, Dr Miriam Poirer, had the following to say to me about the Papadopulos-Eleopulos paper:
Regarding the paper by Papadopulos-Eleopulos et al., I believe that these authors are wrong when they say that AZT does not tri-phosphorylate and incorporate into DNA in AIDS patients because we find AZT incorporated into human DNA of adults, pregnant women and fetuses at birth. (See: Olivero, O.A., Shearer, G.M., Chougnet, C.A., Kovacs, A.A.S., Landay, A.L., Baker, R., Stek, A.M., Khoury, M.M., Proia, L.A., Kessler, H.A., Sha, B.E., Tarone, R.E. and Poirier, M.C.: Incorporation of 3’-azido-3’-deoxythymidine (AZT) into peripheral leukocyte DNA of HIV-1-positive adults and pregnant women, and cord blood leukocyte DNA of infants exposed in utero. AIDS 13: 919-925, 1999. … We believe that AZT would not incorporate if it did not form the tri-phosphate. We are not the first to find AZT incorporated into DNA – but perhaps the first to find it in humans in vivo.
This lawyer presumed to find so much obviously wrong with what the pharmaceutical biochemist was saying (and Poirer too) that he thought it called for a reply:
In their analysis of the molecular pharmacology of AZT, after an extensive analysis of the in vivo triphosphorylation data, Papadopulos-Eleopulos et al comment:
‘Whatever the reason(s), the fact remains that, for AZT to have an anti-HIV effect, it must be triphosphorylated (28), but this is insignificant in vivo. [.....] In their 1986 paper Philip Furman and his research colleagues from the National Cancer Institute, Duke University and Wellcome Laboratories (27) reported that, under ideal conditions, “The IC50 values for the viral reverse transcriptase were 0.7mM with poly(rA).oligo(dT)12-18, and 2.3 mM with activated calf thymus DNA as primer-templates”. In their first clinical trial (28) they acknowledged that “a minimal level for an in vitro antiviral effect” is “above 1mmol/l” of AZTTP. However, such levels of AZT triphosphorylation are not obtained even under ideal, in vitro conditions, and the level of AZT triphosphorylation in vivo is even lower. This means that, as has been generally accepted to date, neither the well known toxic effects of AZT nor any antiretroviral effects can be due to its action as a DNA chain terminator. The question then is, how does AZT produce its toxic effects as well as its anti-HIV effects, if any? Although AZT is not efficiently triphosphorylated it is very efficiently mono-phosphorylated. The mono-phosphorylation of AZT could act as an inhibitor of phosphorylation of cellular constituents, including cellular nucleotides. [.....] It is a well known fact that AZT inhibits mitochondrial DNA (mtDNA) replication. However, since the level of AZT triphosphorylation is negligible, this effect cannot be due to AZT acting as a DNA chain terminator. In their effort to explain the AZT mitochondrial toxicity, researchers from the University of Nagoya studied the mtDNA of mice given either 1 mg/kg/day or 5 mg/kg/day of AZT orally for 4 weeks. Their findings, published in 1991, “suggest that the oxygen damage of mtDNA is the primary cause of mitochondrial myopathy with AZT therapy. oxidative damage of mtDNA can be accumulated during even short period of AZT administration”. They concluded: “The animal model of mitochondrial myopathy with AZT administration reported here seems to be useful for elucidating the mechanism of mtDNA mutations leading to myopathy. However, for AIDS patients, it is urgently necessary to develop a remedy substituting this toxic substance, AZT” (56). [....] The cellular toxicity of AZT was extensively studied by researchers from the State University of New York. In 1996, summarising their findings, they wrote: “Prior to the commencement of the present study, although strong evidence existed that many ddNs, including AZT, could inhibit mtDNA replication, we had not yet substantiated our hypothesis that such inhibition would result in the impairment of oxidative phosphorylation ... Nor had we yet demonstrated a cause-and-effect relationship between the AZT inhibition of mtDNA replication (or its consequence, an impairment of oxidative phosphorylation) on the one hand and an inhibition of cell growth on the other. Thus, the possibility had not been eliminated that AZT was exerting some general cytotoxic effect on the cell, which resulted in an inhibition of cell growth, and this, in turn, was leading to an inhibition of mtDNA replication ... We noticed that the beginning of the AZT-induced inhibitory effect on cell growth occurred at a relatively short time after AZT addition to the medium, a period of time too short to account for the effect to have been brought about by an inhibition of mtDNA replication. This observation led to studies of the early metabolic events that occur upon exposure of the cells to AZT”. In these studies the authors found that: “mitochrondria isolated from cells grown in the presence of pharmacological levels of AZT (5mM) for 5 days and tested for their ability to carry out oxidative phosphorylation showed a marked decrease in ability to synthesize ATP. Further studies of this phenomenon in which the frequency of sampling the medium was in hours rather than days showed early changes in O2 uptake, lactate synthesis, ATP level, and number of mitochondria per cell. Some of these changes, particularly that of ATP level, were observable as early as 3 h after exposure to AZT and, judging from the precipitous decline of the ATP/cell curve between 0 and 3 h, may have begun earlier than that. The 3 h time interval, equivalent to only 7% of the doubling time of the AZT-treated cells, is far too short a period of time to account for the effect brought about by an inhibition of mtDNA replication” (57). In a study published in 1997, researchers from several French institutions “compared the effects of AZT, ddI and ddC on proliferation, differentiation, lipid accumulation, lactate production and mitochondrial enzyme activities in cultured human muscle cells”. They reported that: “All 3 compounds induced a dose-related decrease of cell proliferation and differentiation. AZT seemed to be the most potent inhibitor of cell proliferation. AZT, ddI and ddC induced cytoplasmic lipid droplet accumulations, increased lactate production and decreased activities of COX (complex IV) and SDH (part of complex II)’ (COX=cytochrome c oxidase; SDH=succinate dehydrogenase). Summarising their findings they wrote: “In conclusion, AZT, ddI and ddC all exert cytotoxic effects on human muscle cells and induce functional alterations of mitochondria possibly due to mechanisms other than the sole mtDNA depletion” (58). At present, evidence also exists which shows that AZT is rapidly reduced by compounds containing sulphydryl (-SH); that is, AZT oxidises the -SH groups (59). Ample evidence also exists which shows that oxidation in general (and of -SH in particular) and decreased levels of ATP may lead to many laboratory and clinical abnormalities, including wasting, muscular atrophy, anaemia, damage to the liver and kidney, decreased cellular proliferation, cancer and immunodeficiency (8,19). [...]
In my email to Rasnick, I didn’t include the citations to which Papadopulos-Eleopulos referred. They are:
27. Furman PA, Fyfe JA, St Clair MH, et al. (1986). Phosphorylation of 3í-azido-3í-deoxythymidine and selective interaction of the 5í-triphosphate with human immunodeficiency virus reverse transcriptase. Proc. Natl. Acad. Sci. U S A, 83, 8333-8337.
28. Yarchoan R, Klecker RW, Weinhold KJ, et al. (1986). Administration of 3í-azido-3í-deoxythymidine, an inhibitor of HTLV-III/LAV replication, to patients with AIDS or AIDS-related complex. Lancet, 1, 575-580.
56. Hayakawa M, Ogawa T, Sugiyama S, Tanaka M, Ozawa T. (1991). Massive conversion of guanosine to 8-hydroxy-guanosine in mouse liver mitochondrial DNA by administration of azidothymidine. Biochem. Biophys. Res. Commun., 176, 87-93.
57. Hobbs GA, Keilbaugh SA, Rief PM, Simpson MV. (1995). Cellular targets of 3í-azido-3í-deoxythymidine: an early (non-delayed) effect on oxidative phosphorylation. Biochem. Pharmacol., 50, 381-390.
58. Benbrik E, Chariot P, Bonavaud S, et al. (1997). Cellular and mitochondrial toxicity of zidovudine (AZT), didanosine (ddI), and zalcitabine, (ddC) on cultured human muscle cells. J. Neurol. Sci., 149, 19-25.
59. Handlon AL, Oppenheimer NJ. (1988). Thiol reduction of 3í-azidothymidine to 3í-aminothymidine: kinetics and biomedical implications. Pharm. Res., 5, 297-299.
8. Papadopulos-Eleopulos E. (1988). Reappraisal of AIDS: Is the oxidation caused by the risk factors the primary cause? Med. Hypotheses, 25, 151-162.
19. Papadopulos-Eleopulos E. (1982). A Mitotic Theory. J. Theor. Biol., 96, 741-758.
My reply to Rasnick continued:
In your comment upon my Open Letter, you note:
> AZT and the other DNA chain terminating drugs were designed to be substrates that mimic natural nucleosides that get triphosphorylated and are then added to >growing chains of DNA.
Agreed.
> The purpose of DNA chain termination is to kill the cell.
Agreed.
> That’s just what happens.
The burden of Papadopulos-Eleopulos’s et al meticulous review of all available relevant data is that AZT is not in fact a DNA chain terminator. To repeat, ‘This means that, as has been generally accepted to date, neither the well known toxic effects of AZT nor any antiretroviral effects can be due to its action as a DNA chain terminator.’ They cite Hobbs et al (1995) Cellular targets of 3í-azido-3í-deoxythymidine: an early (non-delayed) effect on oxidative phosphorylation. Biochem. Pharmacol., 50, 381-390: ‘We noticed that the beginning of the AZT-induced inhibitory effect on cell growth occurred at a relatively short time after AZT addition to the medium, a period of time too short to account for the effect to have been brought about by an inhibition of mtDNA replication.’
> For example, below is a recent paper that actually measured AZT incorporated into the DNA of humans taking the drug.
> From O. A. Olivero et al. in their paper entitled ‘Incorporation of zidovudine into leukocyte DNA from HIV-1-positive adults and pregnant women, and cord blood from infants exposed in utero’ (1999) AIDS 13: 919-925:
> ‘We show here that [AZT] is incorporated into leukocyte DNA of most individuals receiving [AZT] therapy, including infants exposed to the drug in utero. Further study of the biological consequences of [AZT]-induced DNA damage in the human population is warranted.’
[When the Olivero] paper came out, Turner pointed out to me its root flaws and described in detail the trouble with the assay that Olivero et al employed. There is no doubt however that AZT, AZT monophosphate and other toxic metabolites accumulate in tissues, including foetal tissues (see below, especially Patterson et al contradicting Olivero et al: ‘Although the active triphosphorylated metabolite was not detected in the fetus, the AZT-monophosphate was detected in almost all fetal tissues examined.’)
With respect, your statement – ‘Whatever efficiency there is of AZT triphosphorylation in vivo, it is certainly sufficient to allow AZT to be incorporated into growing DNA chains’ – appears insupportable. AZT is profoundly toxic to cells for a number of reasons. But they have nothing to do with DNA chain termination. Which is why ‘proviral HIV DNA’ formation is unaffected by AZT, whatever the dose. This is the lynchpin of my action against GlaxoSmith-Kline. […].
I included the Olivero study in my book, not to disingenuously rely on data I believe to be unsound, but to provoke a reaction from Glaxo. Their best argument to refute the Olivero study is that AZT is not triphosphorylated to anything approaching significant levels so cannot bind to mammalian DNA. But that concession would blow its claims to AZT incorporation in ‘retroviral’ DNA out the water.
> DNA chain termination is a serious thing for a cell to experience. It doesn’t require very much incorporation of a DNA chain terminator to kill a cell.
But there has to be significant intracellular AZT triphosphorylation as a prerequisite for AZTTP incorporation to account for the clinically apparent gross cellular toxicity conventionally blamed by AIDS dissidents on cellular DNA chain termination. There isn’t. Would you pass this on too? Because your comment creates the impression that the Olivero paper you cited refutes the heart of the Perth Group AZT critique. It doesn’t. The fallacy that AZT is a DNA chain terminator has to go. It’s akin to convicting the criminal of the wrong crime.
The Patterson study to which I referred in my reply to Rasnick was:
Patterson TA, Binienda ZK, Lipe GW, Gillam MP, Slikker W Jr, Sandberg JA. Transplacental pharmacokinetics and fetal distribution of azidothymidine, its glucuronide, and phosphorylated metabolites in late-term rhesus macaques after maternal infusion. Drug Metab Dispos 1997 Apr;25(4):453-459. Abstract: 3’-Azido-3’-deoxythymidine (AZT) is currently prescribed to pregnant women infected with human immunodeficiency virus to reduce the risk of vertical transmission of the virus to the fetus. Consequently, more information is needed concerning the placental transfer and tissue distribution of AZT and its metabolites. In the present study, the placental transfer and fetal accumulation of AZT, its glucuronide metabolite [3’-azido-3’-deoxythymidine-beta-D-glucuronide (AZTG)], and phosphorylated metabolites were examined at steady-state in near-term rhesus macaques. One to 2 weeks before a chronic infusion, an intravenous bolus of 8 mg/kg AZT was administered to pregnant animals to determine the dose of AZT needed to reach steady-state plasma concentrations. On the day of hysterotomy, the mother was administered an intravenous loading dose of AZT, followed by a 3-hr steady-state intravenous infusion that also included a trace of [3H] AZT. After 3 hr of infusion, the mother was anesthetized, and the fetus was delivered. Plasma and amniotic fluid were analyzed for AZT and AZTG by HPLC, and tissue samples were analyzed for AZT, AZTG, and phosphorylated metabolites by strong anion exchange HPLC. Maternal steady-state plasma concentrations were 1.3-2.2 micrograms/ml for AZT and 2.3-8.0 micrograms/ml for AZTG. Fetal AZT and AZTG plasma concentrations were both lower (0.98-2.3 micrograms/ml and 1.3-5.4 micrograms/ml, respectively) than maternal concentrations, with fetal-to-maternal plasma ratios of 0.63-1.0 for AZT. Fetal tissue distribution of tritium was highest in the kidney and lowest in the brain. Although the active triphosphorylated metabolite was not detected in the fetus, the AZT-monophosphate was detected in almost all fetal tissues examined. Our data indicate that AZT is rapidly converted to the glucuronide and monophosphate metabolites in the fetus after maternal infusion.
This was Turner’s initial criticism of the Olivero paper mentioned in my reply to Rasnick:
Re the Olivero paper (AIDS 1999 13: 919-925): We only looked at the non-pregnant data. These authors measured the number of molecules of AZT incorporated into DNA/one million molecules using an antibody test. They did not use a chemical analysis such as MRI or HPLC. Hell knows why not. Antibodies are fraught with specificity problems. They purchased antibodies to AZT from Sigma and so we know nothing of how these were prepared and tested for authenticity (of what they purport to measure). We do know that drugs given to patients cause the appearance of autoantibodies, that is, antibodies which react with normal cellular (‘self’) components. This includes antibodies to DNA. So we don’t know if the patients taking AZT did not already have antibodies which react with their own DNA regardless of the AZT antibodies. Also, even if the bought antibodies react with AZT in DNA, this does not prove they do not react with other parts of DNA which do not contain AZT. Indeed to prove that the antibodies are specific one would have to spend months comparing the antibody reactions with chemical analyses. Thus there are no data proving the antibody technique they used is specific for what they said they were measuring. A good indication that the reactions are non-specific is:
1. Positive tests were weak (and they varied over three assays).
2. There was no relation between the dose of AZT or the duration of therapy.
3. 1/6 people who had been on AZT in the past reacted (The 1 hadn’t taken any for 2 years and had one of the highest readings (296). 1/6 people who NEVER took AZT reacted. This was 46 molecules which is about the median number for all patients who were on AZT.
Would you trust a test like this?
Rasnick duly circulated my reply, but under another dissent:
I knew I was disturbing a hornets’ nest getting into technical details about AZT. However, I do not accept the Perth Group’s analysis of AZT completely. They still have to explain how AZT gets incorporated into human and animal DNA, both in vitro and in vivo, since AZT must be triphosphorylated for this to happen. See the Olivero et al. paper and references therein. Since AZT is a substrate, and since it only has to get incorporated at very low efficiency for it to kill a cell, and since AZT is prescribed to HIV positive people for life (until recently), and since steady-state enzyme kinetics in vivo typically have substrate concentrations well below their Km values (in contrast to inhibitors which must have their in vivo concentrations well above their Ki values), there is plenty of opportunity for low levels of triphosphorylated AZT to act as a substrate and get incorporated into DNA. The Perth Group’s arguments are not strong enough to change my mind about AZT incorporation into DNA at this point.
It was evident from the quality of this argumentation that I was getting nowhere fast, so I turned our correspondence over to Papadopulos-Eleopulos and Turner, the lead authors of the AZT critique from which I’d quoted extensively in my reply to Rasnick’s demurrer covering my Open Letter to GlaxoSmithKline. They proceeded to demolish the Olivero findings on which Rasnick relied in support of his contention that AZT binds to growing DNA chains and terminates them:
Thank you for asking us to comment on David Rasnick’s criticisms of our AZT paper*. The Olivero et al1 prospective but not blind study was designed to measure the number of AZT molecules per one million nucleotides. In our view there are several problems with Olivero’s et al data and their interpretations including the following: For some unknown reason they did not use a chemical analysis to measure the AZT incorporation. Instead they used a radio-immunoassay based upon a polyclonal anti-AZT antibody. It is well known that antibodies, including monoclonal antibodies react with antigens (molecules) other than the ones one would like.2-9 Indeed, there are instances where ‘cross-reactive antigen binds with higher affinity than the homologous antigen itself ... The most obvious fact about cross-reactions of monoclonal antibodies is that they are characteristic of all molecules and cannot be removed by absorption without removing all reactivity ... Even antigens that differ for most of their structure can share one determinant, and a monoclonal antibody recognizing this site would then give a 100% cross-reaction. An example is the reaction of autoantibodies in lupus with both DNA and cardiolipin’. However, ‘It should be emphasised that sharing a ‘determinant’ does not mean that the antigens contain identical chemical structures, but rather that they bear a chemical resemblance that may not be well understood, for example, a distribution of surface charges’.9 In the Olivero paper no data exist, in fact no mention is made in regard to the specificity of the antibody test, that is, if a one to one correlation existed between the number of AZT molecules per 106 nucleotides as determined by their method and by a chemical method. However, discussing their findings they wrote: ‘It is possible that the antiserum might also recognise ZDV attached to proteins, and it is certain that free ZDV will be recognised’. Although they claim to have used ‘stringent methods of DNA purification’, they did not exclude the possibility that proteins may have been present in their DNA preparation. Furthermore, if the antibodies react with proteins, then it is at least as likely that they may also react with parts of DNA not containing AZT. In this regard, it is highly important to note that although ‘samples from ZDV-treated individuals (86% from non-pregnant adults, 66% from pregnant women, and 68% from infants) had measurable ZDV-DNA levels by ZDV-RIA’, so did 2 out of 12 (17%) non-pregnant adults, used as controls, one of them having one of the highest values (296.3 ± 36.5 molecules ZDV/106 nucleotides) detected in any individual. Since neither Olivero, nor anybody else has proven the test used in this study is specific, it is not possible to draw any conclusion in regard to AZT incorporation into DNA.
1. Olivero OA, Shearer GM, Chougnet CA, et al. Incorporation of zidovudine into leukocyte DNA from HIV-1-positive adults and pregnant women, and cord blood from infants exposed in utero. AIDS 1999;13:919-25.
2. Guilbert B, Fellous M, Avrameas S. HLA DR specific monoclonal antibodies cross react with several self and nonself non MHC molecules. Immunogenetics 1986;24:118 121.
3. Pontes de Carvalho LC. The faithfullness of the immunoglobulin molecule: can monoclonal antibodies ever be monospecific? Immunol. Today 1986;7:33.
4. Ternynck T, Avrameas S. Murine natural monoclonal antibodies: a study of thier polyspecificities and their affinities. Immunol. Rev. 1986;94:99 112.
5. Owen M, Steward M. Antigen recognition. In: Roitt I, Brostoff J, Male D, ed. Immunology. 4th ed. London: Mosby, 1996: 7.1 7.12.
6. Gonzalez Quintial R, Baccala R, Alzari PM, et al. Poly(Glu60Ala30Tyr10) (GAT) induced IgG monclonal antibodies cross react with various self and non self antigens through the complentarity determining regions. Comparison with IgM monoclonal polyreactive natural antibodies. Europ. J. Immunol. 1990;20:2383 2387.
7. Parravicini CL, Klatzmann D, Jaffray P, et al. Monoclonal antibodies to the human immunodeficiency virus p18 protein cross react with normal human tissues. AIDS 1988;2:171 177.
8. Fauci AS, Lane HC. Human Immunodeficiency Virus (HIV) Disease: AIDS and Related Disorders. In: Isselbacher KJ, Braunwald E, Wilson JD, Martin JB, Fauci AS, Kasper DL, ed. Harrison’s Principles of Internal Medicine. 13th ed. New York: McGraw Hill Inc., 1994: 1566 1618.
9. Berzofsky JA, Berkower IJ, Epstein SL. Antigen Antibody Interactions and Monoclonal Antibodies. In: Paul WE, ed. Fundamental Immunology. 3rd ed. New York: Raven, 1993: 421 465.
*Papadopulos-Eleopulos E, Turner VF, Papadimitriou JM, Causer D, Alphonso H, Miller T. A critical analysis of the pharmacology of AZT and its use in AIDS. Current Medical Research and Opinion 1999;15 (Supplement 1):1s-45s.
As I’d requested, Rasnick circularised this response from Papadopulos-Eleopulos and Turner, but did so under this statement:
I am posting by request the Perth Group response to my last comments on the incorporation of AZT into DNA.
We have all had our say regarding the incorporation of AZT into DNA. I agree with my friend Sam Mhlongo that: “Our struggle against mainstream orthodoxy is beyond medical science alone but encompasses the following factors: Social Political Economic. Our view is that the battle will not be won on hard medical science alone.” Therefore, I end my contribution to the technical details of AZT incorporation. I return to the political and sociological front.
To be frank I was staggered. Apart from being a non-sequitur, his answer suggested to me a deliberate unwillingness to consider the opposing case. A startling attitude, I thought, by a pharmacologist who billed himself as AZT expert, and who had testified in court as such. Nonetheless, I then suggested to the Perth Group that their response merited amplification, and they responded thus:
Many thanks for pointing out we did not answer David Rasnick’s theoretical argument, that even very low levels of triphosphorylated AZT may lead to ‘AZT incorporation into DNA’ resulting in inhibition of DNA synthesis and thus cell killing (toxicity). There were two reasons for this:
(i) It is a theoretical argument;
(ii) Unless there is some indication that AZT is incorporated, into DNA, at any level, we wonder what relevance attaches to such theoretical ruminations.
In our view we can do no better in answering this then by quoting from a 1995 study by researchers from the University of Colorado. ‘In assays containing 10mM dNTPs, AZTTP poorly inhibited DNA synthesis in nuclei obtained from K562 and CEM cells, with IC50 values greater than 500mM. The presence of 1mM AZTMP slightly enhanced inhibition in K562 nuclei, but the AZTTP concentration needed for significant inhibition remained several hundred-fold higher than the AZTTP concentration reported in AZT-treated cells (about 1mM) ... These results suggest that inhibition of nuclear DNA replication is unlikely to account for the side effects associated with AZT therapy’. {Yan et al, J Biol Chemistry 1995; 270:22836-22841}.
Since even very high levels of AZTTP do not inhibit DNA synthesis, it is not possible for the picomolar concentrations present in patients given AZT to inhibit DNA synthesis. This means that AZTTP is not incorporated into DNA and thus it cannot act as DNA ‘chain terminator’. This applies to the synthesis of both cellular and HIV DNA.
Rasnick refused to circulate this perfected answer by the Perth Group to his criticisms of their conclusion that AZT is not incorporated into DNA and cannot act as a DNA chain terminator, and said so in the following terms:
I’m done with this subject. If the Perth Group is so committed to their views they should write a proper scientific paper and submit it for publication in a journal. Duesberg has said that he will use whatever influence he has to get their paper published. These email technical debates are pointless and time consuming. If the Perth Group publishes their arguments in a journal, I will be more inclined to continue to challenge their claims. But not now. One final thing. Why does the Perth Group seem to prefer to go through second parties? Why don’t they present things directly? Why don’t they publish? Duesberg and I have difficulty publishing, but we do get published.
I was struck by the inappropriateness of Rasnick’s hostile tone in this purely scientific disagreement, and amazed that he should be unaware that the Perth Group’s AZT critique had indeed been published, and in a very estimable medical journal too – notwithstanding that I had quoted from it extensively in my reply to his initial dissent covering my Open Letter, and that it had been discussed in Nature. It was also apparent that Rasnick was ignorant of the Perth Group’s extensive publishing record. And the Perth Group were answering my enquiries, hence my intermediary role. By trivialising as ‘pointless’ and ‘technical’ a crucial discourse about the basic pharmacology of AZT, Rasnick was passing up an opportunity to kick the chair out from under it, by vitiating its basic warrant as a medicine: the claim that it terminates DNA, and proviral HIV DNA in particular. The obvious lesson from the withdrawal of the Threakall action in England and the failure of the Tyson defence in the US is that it’s no good founding cases on AZT’s toxicity alone without impeaching its efficacy, because doing so leaves the door open to GSK to claim a benefit outweighing the risk, to which the only answer is an attack on the HIV-AIDS construct, a route that widens the issues to inconveniently broad proportions. I decided to call off the exchange.
Dr Peter Moore, GlaxoSmithKIine’s South African medical director, eventually answered my Open Letter as follows:
The paper by Furman et al described an in-vitro assay (exogenous HIV-1 RT assay) to demonstrate that ZDV-triphosphate is a selective inhibitor of reverse transcriptase from HIV-1, relative to polymerase alpha from mammalian cells. This assay was therefore used to establish that zidovudine had the potential to inhibit HIV-1 at concentrations that would not inhibit mammalian cells. It is not appropriate to pick one value from an initial in-vitro experiment and ignore all later studies, including those in HIV positive subjects, that have take account of increasing knowledge and experience in the scientific community.
It is known that there is a poor correlation between ZDV phosphorylation (and other NRTIs used in clinical practice) and antiretroviral activity, due to both methodological issues and with the techniques employed and other factors such as differential activation depending on the cell type and differential transport of the drug into or within varying cell types. These issues are confirmed in recent reviews of the subject by independent experts in the field.
· Hoggard PG, Back DJ, Nandwani R. Intracellular nucleoside analogue phosphorylation and antiretroviral therapy strategies. Journal of HIV Therapy 1998;3(4):81-6
· Peter K, Gambertoglio JG. Intracellular phosphorylation of zidovudine and other nucleoside reverse transcriptase inhibitors used for human immunodeficiency virus (HIV) infection. Pharmaceutical Research 1998;15(6):819-25
· Pastor-Anglada M, Felipe A, Casadro FJ. Transport and mode of action of nucleoside derivatives used in chemical and antiviral therapies. Trends in Pharmacological Sciences 1998;19(10): 424-30
Since the early identification of NRTIs with anti-HIV activity, the reverse transcriptase activity has been well characterised at the molecular and enzymatic level. It is clear that the major activity of NRTIs is by chain termination.
· Jonckheere H, Anne J, De Clercq E. The HIV-1 Reverse Transcription (RT) Process as Target for RT Inhibitors. Medicinal Research Reviews 2000;20(2):129-54
A wide-ranging analysis of the major trials of NRTI mono- and dual therapy, including Concorde, recently showed that ZDV monotherapy led to a reduction in disease progression, however this was not sustained. This is unfortunately the case for all monotherapy with any HIV drug due to the rapid development of resistance.
· Zidovudine, didanosine, and zalcabitine in the treatment of HIV infection: meta-analyses of the randomised evidence. HIV Trialists’ Collaborative Group. Lancet 1999;353(9169);2014-25
Reviews of ZDV have clearly shown that the drug has time-limited efficacy in monotherapy and is an effective component of triple combination therapy, evidenced by the inclusion of the drug in treatment guidelines for all major countries with experience in combating the HIV epidemic (International and UK cited below).
· Wilde MI, Langtry HD. Zidovudine. An update of its pharmacodynamic and pharmacokinetic properties, and therapeutic efficacy. Drugs 1993;46(3):515-78
· Carpenter CC, Cooper DA, Fischl MA, Gatell JM, Gazzard BG, Hammer SM et al. Antiretroviral therapy in adults: updated recommendations of the International AIDS Society-USA. JAMA 2000;283(3):381-90
· BHIVA Executive Committee. British HIV Association (BHIVA) guidelines for the treatment of HIV-infected adults with antiretroviral therapy. HIV Medicine 2000;1:76-101
Moore’s letter was a thrill to read. The prospect of cross-examining him (or some stand-in stooge) on his statements over several days, with journalists from the world’s leading newspapers and news organisations (all listed in my copy of the Foreign Correspondents Directory) watching them dismantled, was something to live for.
The reply I sent to noseweek for forwarding to GlaxoSmithKline dealt only with Moore’s most conspicuously stupid misstatements:
AD PARAGRAPH 2
Actually, the purpose of the Furman study wasn’t to describe an assay as you claim; it was to investigate the relative selectivity of AZT triphosphate for the enzymes you mention. (Their findings in this regard were disproved in a slew of subsequent studies: Genetica 95: 103-109, 1995.) But the relevant bit from their paper was: ‘IC50 (concentration of inhibitor that inhibits enzyme activity 50%) values were determined for azidothymidine triphosphate with HIV reverse transcriptase…’ Since the Furman study determined the all-important IC50 value of AZTTP, it was entirely ‘appropriate’ to ‘pick’ it. There aren’t any other ‘later studies’ in vitro reporting lower IC50 values for AZTTP that I ‘ignored.’ And not a single one in vivo for ‘HIV positive subjects’. No doubt because apart from the first one which everyone agrees Toyoshima et al botched, all the studies to which I referred you, ‘increasing knowledge and experience in the scientific community’, have reported that human cells in vivo triphosphorylate AZT to only miniscule concentrations – far lower than the minimum IC50 value for AZTTP that Furman et al ascertained in vitro for the activated drug to have antiretroviral activity. You can’t waffle your way out of this. You’ve got serious trouble coming and you know it. The kind that Big Tobacco is having in American courts right now.
AD PARAGRAPH 3
Certainly, due to saturation and the other factors that you mention, there are poor correlations between the size of the AZT dose administered and intracellular concentrations of AZT, AZT mono-, di-, and triphosphate. But it is vain to suggest that there is a poor correlation between ‘AZT [tri-]phosphorylation and antiviral activity’. Because unless AZT is triphosphorylated to its IC50 level, it can’t be antiretroviral. And the more the merrier. But no matter what the dose given, the ‘later studies’ I cited have consistently shown that the cells of patients given AZT cannot triphosphorylate it to levels anywhere near its minimum effective concentration.
AD PARAGRAPH 4
Furman et al speculated: ‘Incorporation of azidothymidylate [AZTTP] into a growing [proviral HIV] DNA strand should terminate DNA elongation and thus inhibit DNA synthesis.’ And thus block HIV replication. That was the idea (which you assert as an established fact). In reality we know AZT doesn’t terminate viral DNA chain synthesis in vivo, because as I explained, it doesn’t reduce HIV DNA levels i.e. ‘viral burden’, or any other direct marker for HIV infection levels. This is because AZT is not triphosphorylated significantly in the cells of people taking it, evident since the early nineties. And unless the drug is triphosphorylated, it can’t be incorporated into growing viral DNA chains to terminate them. In short, taken as a medicine, AZT can’t and doesn’t work as claimed. Indeed your own Product Information advisory concedes: ‘The relationship between in vitro susceptibility of HIV to [AZT] and the inhibition of HIV replication in humans or clinical response to therapy has not been established.’ Which is another way of saying that you’ve never proved AZT effective as an antiviral medicine. Well, precisely.
The question you are going to have to answer sooner or later is this: Knowing that AZT was synthesized in 1961 as a cell poison, why did you commence marketing it as an ‘antiretroviral’ drug in 1987 before proving that it has this latter activity in vivo? And why have you disregarded all studies published since, indicating that it doesn’t. Especially knowing how harmful it is.
AD PARAGRAPH 5
Like all indiscriminate cell poisons such as arsenic and mercury salts in the olden days, AZT might conceivably allay certain infections temporarily, due to its established bactericidal activity. But it can’t be and isn’t antiretroviral for the reasons I’ve set out. You haven’t addressed the fact that the biggest and best-conducted AZT trial yet conducted, the Concorde trial, found no therapeutic benefits for asymptomatic HIV-positive patients, and the extended results of the trial found that it increases rather than reduces risk of death. And as you know, numerous other clinical trials conducted independently and not funded by your company have also reported that AZT is at best useless, and at worst accelerates clinical decline and death. Obviously, excuses about ‘development of resistance’ for its clinical inefficacy don’t cut it unless you have established that AZT has some initial anti-HIV activity in vivo. You haven’t.
AD PARAGRAPH 6
It may be that AZT remains the biggest selling AIDS drug (which, together with 3TC, hauled in $1.1 billion last year), and is still recommended by the ‘AIDS experts’ as part of ‘triple combination therapy’ notwithstanding its manifest inefficacy and toxicity. Your drug-marketing budget of $4.7 billion last year alone might have something to do with it. But widespread use doesn’t establish it works, any more than the standard textbook administration of arsenic to scurvy-ridden English sailors and soldiers did. Anyway, facing a mountain of severe toxicity and clinical inefficacy reports, the US Department of Health and Human Services has recently done an about-face on its ‘hit early, hit hard’ treatment recommendations, and on 6 February this year laid down new guidelines urging that the administration of AZT and other ‘antiretrovirals’ be delayed. And as we speak, the British HIV Association is preparing its own treatment guidelines, proposing an even longer delay in the initiation of treatment.
Quoted in Esquire in April 1999 leading US AIDS clinician Dr Michael Saag at the University of Birmingham, Alabama remarked that doctors ‘should expect failure with whatever [‘antiretroviral’ cocktail they] first use. We should plan on it. We should prepare for it. Clinicians should expect failure’. He noted that instead of getting better, his patients on ‘antiretroviral’ cocktails were dying: ‘They aren’t dying of a traditionally defined AIDS illness … I don’t know what they’re dying of, but they are dying. They’re just wasting and dying.’ The logic may be elusive to the ‘AIDS experts’, but gee, could it be that cell poisons poison cells?
GlaxoSmithKline has never pertinently addressed the triphosphorylation problem. When in the late nineties Gerrit Brand (mentioned earlier) taxed GSK in the Netherlands about it, the company responded by sending him a Product Information sheet containing the usual exposition of its DNA chain termination model. The interesting bits, set out under the heading Published Information: Phosphorylation of Zidovudine, are set out below, broken up for a running commentary, with the authority cited by GSK for each of its claims plugged in by me as we go:
ZDV-TP remains present inside the cell for 12 hours to 24 hours following incubation of CCRF-CEM cells with ZDV
· Fridland A, Connelly M, Ashmun R. Relationship of deoxynucleotide changes to inhibition of DNA synthesis induced by the antiretroviral agent 3’-azido-3’-deoxythymidine and release of its monophosphate by human lymphoid cells (CCRF-CEM). Mol Pharmacol 1990;37:665-70
and following steady state dosing with ZDV in patients.
· Barry M, Khoo S, Veal G, et al. The effect of zidovudine dose on the formation of intracellular phosphorylated metabolites. AIDS 1996;10:1361-7
You’d think from this sentence that the company meant AZTTP is found in patients’ cells at significant and not merely trace levels.
The amount of ZDV-TP formed is 25 to 160 fold the amount necessary to bind to reverse transcriptase.
· Furman P, Fyfe J, St. Clair M, et al. Phosphorylation of 3’-azido-3’-deoxythymidine and selective interaction of the 5’-triphosphate with human immunodeficiency virus reverse transcriptase. Proc Nat’l Acad Sci 1986;83:8333-7
This statement is intended to clinch it. The trouble is that the Furman et al study, cited as authority for this proposition, was conducted in vitro, in ideal artificial conditions. But what goes on in the body is another matter altogether. As revealed by the sixteen studies cited in my Open Letter, which find ‘the amount…of AZTTP formed’ in vivo to be nowhere near ‘the amount necessary to bind to reverse transcriptase’ in order to inhibit HIV RNA retrotranscription to a meaningful extent.
Furthermore, this amount of ZDV-TP is known to be clinically sufficient as monotherapy with Retrovir has been shown to significantly reduce the mortality rate and the incidence of opportunistic infections in patients with AIDS
· Fischl M, Richman D, Grieco M, et al. The efficacy of azidothymidine (ZDV) in the treatment of patients with AIDS and AIDS-related complex. N Engl J Med 1987;317:185-91
This is rather hard to reconcile with the AZT ‘Product Information’ advisory, quoted earlier: ‘The relationship between in vitro susceptibility of HIV to [AZT] and…clinical response to therapy has not been established.’ We discussed the corruption of the Phase II AZT clinical trial in ‘Licensing AZT’. Nobody nowadays, with one hand on the Bible and the other aloft, would claim that AZT monotherapy saves lives. Citing the Fischl paper to pretend that it does is like citing a paper in Lancet from the 1800s vaunting the discovered benefits of arsenic for scurvy. Even GlaxoSmithKline’s Dr Peter Moore agrees: during the Carte Blanche television programme The AZT Debate on 7 November 1999, he stated: ‘The days of using AZT as a monotherapy are gone.’ But that’s not what the package insert says. That the drug has proved to be useless. Unless made to work somehow by being mixed with something else. As Dr Arthur Gottlieb in the US agreed in an article in the Rocky Mountain News in which he was quoted on 3 May 1998: ‘…everybody will tell you that AZT is not very good. If you prescribe it as a single drug to a patient, it would be deemed to be malpractice.’
As for the claim, ‘this amount of ZDP-TP is known to be clinically significant’, we lawyers refer to such as a fraudulent misrepresentation. Its gravamen is that AZT is triphosphorylated sufficiently in the body to exert a significant antiviral effect. It isn’t.
But then, under the subheading, ‘Pharmacologic Significance of Zidovudine Phosphorylation’, GSK cautions: ‘It should be noted that in vitro activity does not necessarily correlate with clinical response.’ This is the first hint of something remiss. A truthful statement for a change. But in abstract, so you don’t know what they mean. And ought to spell out. Remember my Open Letter: every single study of the effect of AZT administration on what ‘AIDS experts’ call HIV DNA (viral burden) has revealed that the drug does not reduce it. That is, AZTTP is not being incorporated in ‘HIV DNA’ and is not terminating it. In other words it doesn’t work as claimed. This statement obliquely concedes that whatever is going on in test tubes cannot simply be extrapolated to the real world, i.e. to the cells of our bodies. But so indirectly, and set against a background of so many misstatements, that you’d never think so.